Screening with Insertional Libraries
We have generated transposon-insertion libraries of plasmids, with each plasmid carrying an insert of yeast genomic DNA containing a single random insertion of a bacterial transposon. This transposon-mutagenized library is extremely useful as a biological reagent; the protocols presented here describe the application of this library for expression profiling and phenotypic screening in yeast.
Briefly, library DNA is introduced into an appropriate strain of yeast by standard methods of DNA transformation. By homologous recombination, the transposon-mutagenized genomic DNA will replace its chromosomal locus, generating a yeast strain carrying a chromosomal transposon insertion. Transformants carrying an in-frame fusion of this transposon to yeast protein-coding sequence may be identified using a color assay for β-galactosidase ( β-gal) activity (the lacZ reporter is encoded within the specially designed transposon). The resulting bank of yeast strains containing productive lacZ fusions may then be screened for any desired mutant phenotype.
Amplifying Library DNA in E. coli
Transforming Yeast
Screening for β-galactosidase Activity
Identifying Insertion Sites by Vectorette PCR
Amplifying Library DNA in E. coli
Aliquots of library DNA are distributed in 10 pools as dried-down solutions, which may be suspended in an appropriate volume of TE buffer, pH 8.0 (e.g., at a concentration of approximately 100 ng/μl). The plasmid-based library was generated in a vector carrying tetracycline and kanamycin resistance.
- Introduce a suitable amount of insertion library DNA into any tetracycline- and kanamycin-sensitive E. coli strain by standard transformation procedures. Select transformants on LB medium supplemented with tetracycline (3 μg/ml) and kanamycin (40 μg/ml) using plates 14 cm in diameter. Approximately 10,000 transformants should be obtained per pool (approximately 100,000 in total) following overnight growth at 37°C.
- Elute transformant colonies as follows: place 6 ml of LB medium onto each plate and scrape cells into a homogenous suspension. Dilute an aliquot of this eluate into LB medium supplemented with tetracycline (3 μg/ml) and kanamycin (40 μg/ml) to yield a culture of nearly saturated cell density. Incubate at 37°C with aeration for 2-3 hours.
- Isolate plasmid DNA by any standard miniprep or large-scale protocol.
Transforming Yeast
Yeast transformations are performed using a modification of the lithium acetate/single-stranded DNA/polyethylene glycol (PEG) method described by Chen and colleagues.
- Digest a small aliquot of plasmid DNA (e.g., 1 μg) with NotI. Subsequently, analyze a portion of the reaction mixture by agarose gel electrophoresis to ensure release of mTn-mutagenized yeast DNA from the plasmid vector. Upon electrophoresis, a distinct 2.1-kb band (corresponding to the vector) and broad 8-kb band should be visible: the broad 8-kb band consists of 2-3-kb inserts of yeast genomic DNA carrying the 6-kb mTn construct. Store the remaining reaction mixture for later use in step 4.
- 2. Grow a 10-ml culture of any desired ura3 yeast strain to mid-log phase (a density of 107 cells/ml or OD600 of approximately 1) maintaining appropriate selection if applicable. Ideally, choose a diploid yeast strain to screen for desired patterns of gene expression. To screen for disruption phenotypes, a haploid strain is often used; from previous studies (S. A. des Etages and M. Snyder, unpublished), we estimate that 10% of transposon insertions in essential genes are viable. For the eventual analysis of HAT-tagged proteins, choose a ura3 leu2 strain.
- 3. Pellet cells in a clinical tabletop centrifuge at 1100xg for 5 minutes. Wash once with 5 volumes of One-Step Buffer [0.2 M lithium acetate, 40% (w/v) PEG 4000, 100 mM 2-mercaptoethanol].
- 4. Resuspend cells in 1 ml One Step Buffer supplemented with 1 mg denatured salmon sperm DNA. Add 100-μl aliquots from this suspension to 0.1-1 μgNotI-digested plasmid DNA from step 1. Use a small quantity of transforming DNA in order to minimize generation of transformants containing more than one insertion. Vortex, and incubate at 45°C for 30 minutes.
- 5. Pellet cells and subsequently suspend in 400 μl SC -Ura medium. Spread 200-μl aliquots onto SC -Ura plates, and incubate at 30°C for 3-4 days. Up to 1000 transformants may be recovered per ?g of transforming DNA. To ensure 95% coverage of the genome (without regards to in-frame reporter activity), screen 30,000-50,000 colonies. To identify in-frame insertions within at least 95% of all yeast genes, screen approximately 180,000-200,000 transformants for β-gal activity.
Screening for β-galactosidase Activity
Yeast transformations are performed using a modification of the lithium acetate/single-stranded DNA/polyethylene glycol (PEG) method described by Chen and colleagues.
- To maximize detection of lacZ fusions expressed at low levels, patch transformant colonies onto YPD plates (supplemented with 80 μg/ml adenine if using an ade2 host strain) at a density of up to 100 colonies per plate.
- 2. Place a sterile disc of Whatman 3MM filter paper (Clifton, NJ) onto a plate of SC -Ura medium; repeat for as many plates as needed. Replicate transformant cells onto filter-covered plates and incubate overnight at 30°C. Alternative growth conditions (e.g., growth on sporulation medium) may be substituted as desired.
- 3. Following overnight growth, lift filters from plates and place in the lid of a 9-cm glass petri dish. Place this lid inside a closed 15-cm petri dish containing chloroform. Incubate for 10-30 minutes.
- 4. Place filters colony-side up onto fresh X-gal plates [5-bromo-4-chloro-3-indolyl-βD-galactopyranoside (x-Gal, 120 μg/ml),0.1 M NaPO4 (pH 7.0), 1 mM MgSO4 in 1.6% (w/v) agar]. Incubate inverted at 30°C for up to 3 days. After several days of growth, β-gal levels can be reliably estimated from the observed intensity of blue staining. We typically observe β-gal activity in 12-16% of transformants.
Transformants containing in-frame lacZ-fusions may be screened for mutant phenotypes simply by incubating these mutants under desired growth conditions (e.g., in the presence and absence of a particular drug of interest).
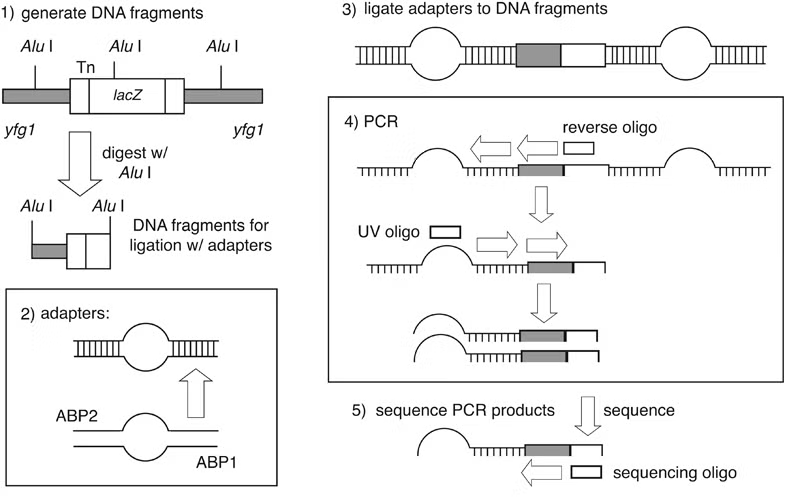
Identifying Insertion Sites by Vectorette PCR
Genomic sites of transposon insertion may be identified within strains of interest by several approaches. Genomic DNA immediately adjacent to a transposon insertion can be recovered in E. coli by plasmid rescue. Alternatively, insertion sites may be identified through direct genomic sequencing of mTn-mutagenized strains using a transposon-specific primer. PCR-based techniques are also applicable as a means of detecting transposon insertions: most notably, the vectorette PCR method of Riley et al. has been successfully utilized for this purpose.
In vectorette PCR, genomic DNA is digested with a blunt-end restriction endonuclease possessing a 4-6 base pair recognition sequence. Digested DNA fragments are ligated to a pair of annealed primers containing a non-homologous central region; these primer pairs form “anchor bubbles” flanking each genomic fragment. PCR is then performed using a primer complementary to the transposon and a primer identical to sequence within the anchor bubble. During the initial round of amplification, only the mTn primer can bind its template; however, during subsequent cycles, the anchor bubble primer can anneal to the extended mTn primer, resulting in selective amplification of DNA sequence adjacent to the point of transposon insertion.
The vectorette PCR protocol provided below should yield approximately 200-400 ng of product, constituting sufficient template for 2-3 sequencing reactions.
- Prepare genomic DNA by any standard protocol (e.g., the Zymolyase-based method of Philippsen and colleagues); care should be taken to obtain high-quality DNA, as this is critical to successful PCR amplification. Digest 5 μg of yeast genomic DNA with a blunt-end restriction endonuclease (such as AluI) in a total volume of 20μl. After overnight digestion, the enzyme is heat inactivated by incubating 20 minutes at 65°C.
- Primers ABP1 and ABP2 (please see figure) are annealed to each other to form the adaptor anchors by mixing 1 pmole of each primer in 200 μl of annealing buffer containing 10 mM Tris, 10 mM MgCl2, 50 mM NaCl. The primer mixture is heated for 5 minutes at 95°C and allowed to slowly cool to 37°C.
- The adaptors are ligated to the DNA fragments by adding 1 μl of the annealed primers, 0.25 μl of 10 mM ATP, 3 μl of 10x restriction buffer used in the digest, and 24.25 μl H2O to the 20 μl restriction digest mixture from Step 1. The ligation reaction is incubated overnight at 16°C.
- A standard 100 μl PCR reaction is set up using 5 μl from the ligation mixture, 2.5 μl each of primers UV and M13(-47) (please see figure) at 20 mM, 5 U of Taq polymerase and 1 μl of dNTP’s (at 20 mM each dNTP) in a final volume of 100 μl. The PCR program consist of one cycle of 2 minutes at 92°C, followed by 35 cycles of 20 seconds at 92°C, 30 seconds at 67°C and 45 to 180 seconds at 72°C with a final extension of 90 seconds at 92°C.
- Each PCR product is gel purified using standard protocols into a final volume of 30 μl TE. 10 μl of the purified product is sufficient for one sequencing reaction.