

We use cancer cell lines and budding yeast to study lysosomes.
The lysosome (or vacuole in fungi and plants) is an essential organelle responsible for the digestion and recycling of intracellular materials delivered by endomembrane trafficking and autophagy. It also plays an important role in intracellular signal transduction by regulating the localization and activity of the mTOR signaling complex. Not surprisingly, lysosome dysfunction leads to ~ 70 different lysosomal storage diseases (LSDs) and contributes to aging-related neurodegenerative, muscular dystrophy, and cardiovascular diseases.
We are fascinated by everything related to the lysosome. In our lab, we want to understand how it is born(biogenesis) and how it is regulated to stay functional (quality control). We are also curious about how lysosome communicates with other organelles. Currently, we are working on the following questions using budding yeast, human cancer cell lines, and vertebrate animal models:
Lysosome Membrane Quality Control through ubiquitin and ESCRT-dependent microautophagy
The lysosomal membrane has hundreds of proteins essential for organelle function. For example, lysosomes contain many nutrient transporters and ion channels to move materials across the membrane. Lysosome membranes also contain Rabs, SNAREs, and tether proteins to receive cargoes delivered by late endosomes and autophagy. Despite their importance, very little is known about their regulation mechanisms. We also don’t know how damaged or mutated lysosomal membrane proteins are removed.

Using budding yeast as a model system, we discovered that lysosomal membrane proteins are regulated by E3 ubiquitin ligases in response to environmental cues. For example, removing lysine from the growth media will lead to the ubiquitination of a vacuolar lysine importer Ypq1(Fig.1). The ubiquitination is carried out by a vacuolar E3 ligase complex Ssh4-Rsp5. Afterward, Ubiquitinated Ypq1 will be internalized by the ESCRT machinery into the vacuole lumen and degraded. We termed this process the ubiquitin- and ESCRT-dependent microautophagy. We also demonstrated this pathway is conserved from yeast to humans.
My lab discovered three independent lysosomal E3 ligase systems in yeast, including Ssh4-Rsp5, Dsc complex, and Pib1. In humans, there are over a dozen lysosomal E3 ligases, most of which have not been characterized.
The current focus of this project includes 1) understanding how yeast vacuolar E3 ligases selectively recognize their membrane substrates, 2) characterizing human E3 ligases with a focus on identifying their membrane substrates, 3) purifying and reconstituting lysosomal membrane transporters into proteoliposomes for functional studies.
Characterization of TMEM251 that causes a severe lysosome storage disease in humans
In a genome-wide CRISPR-Cas9 knockout screening to identify critical genes for human lysosome function, we unexpectedly discovered TMEM251, a gene of unknown function. At the cellular level, we observed that lysosome numbers are remarkably upregulated after knocking out TMEM251. However, these lysosomes are defective, as evidenced by the numerous undigested materials within their lumen (Fig.2A-B). In humans, TMEM251 mutations lead to severe genetic disease. Patients suffer from severe skeletal dysplasia, protruding abdomens, and coarse facial features. High levels of lysosomal enzymes can also be detected in their plasma. But the molecular mechanism of the pathogenesis was unknown.

Further studies in our lab uncovered that TMEM251 plays an essential role in the biogenesis of the M6P pathway. TMEM251 gene encodes a small membrane protein that is enriched in the Golgi (Fig.2C). Without TMEM251, most lysosomal enzymes lose their M6P modification and can no longer traffick to the lysosome. These enzymes either accumulate at the Golgi or are secreted out of the cell (Fig. 2D). Why is the M6P pathway defective? It turns out that TMEM251 is very critical for the stability of N-acetylglucosamine-1-phosphotransferase (GNPT), the Golgi enzyme responsible for adding M6P to lysosomal enzymes. Without TMEM251, most GNPT disappeared. And the only GNPT form that is left is the unprocessed precursor. Not surprisingly, the disease phenotypes of TMEM251 patients resemble GNPT patients. Because of that, we named the disease Mucolipidosis Type V.
The follow-up studies include 1) understanding the relationship between TMEM251 and GNPT, aka, why does knocking out TMEM251 lead to GNPT instability? 2) generating TMEM251 KO Zebrafish model to understand the importance of TMEM251 in vertebrate development. These KO fish have severe skeletal dysplasia, defective cartilage development, and enormous heart and abdomen, all consistent with patients’ phenotypes. But why?
The role of the ERAD pathway in the pathogenesis of Cystinosis
Cystinosis is a recessive lysosome storage disease caused by mutations in cystinosin, the cystine exporter on the lysosomal membrane. Cystinosin mutations will lead to a massive buildup of cystine within the lysosomal lumen, eventually impairing the entire lysosome function. In untreated patients, the kidneys and eyes are the first organs to be affected. The disease progression leads to complete renal failure within the first ten years of age. Patients also suffer from hypothyroidism, weakened bones and muscles, and reduced cognitive abilities.
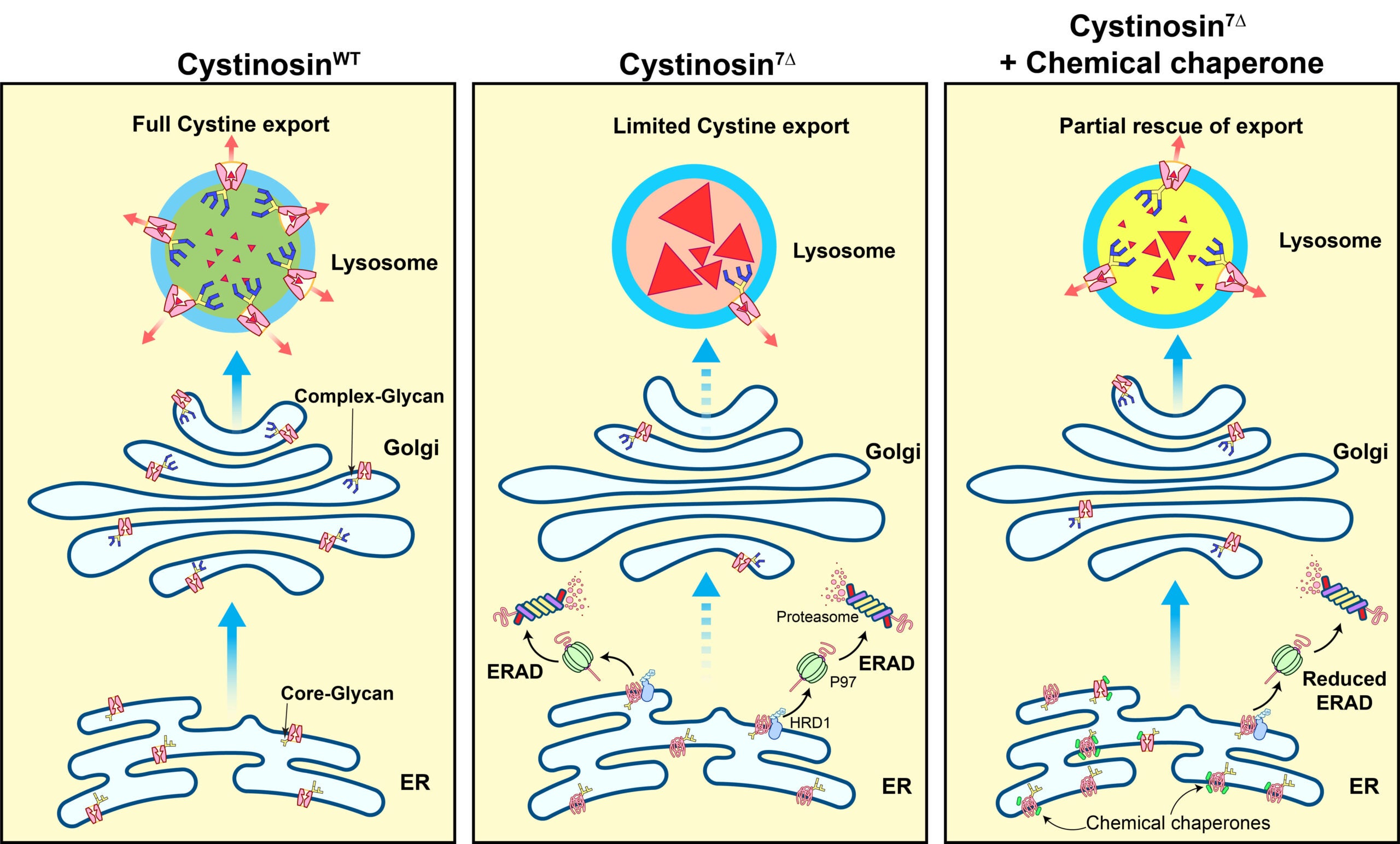
We study the pathogenesis mechanism of Cystinosis with the hope of developing new treatment strategies. Our central hypothesis is that some cytosine mutations might affect protein stability and lead to premature degradation. As proof of principle, we studied the degradation mechanism of Cystinosin7Δ, a mutant that harbors a deletion of seven amino acids (I,T,I,L,E,L,P) from positions 67 to 73. We demonstrated that Cystinosin7Δ is trapped at the ER and quickly degraded by the ER-associated degradation pathway. Importantly, we showed that some chemical chaperones that assist membrane protein folding at the ER could be used to treat Cystinosin7Δ, resulting in a dramatic reduction(~70%) of cystine levels in patient fibroblasts.
The follow-up studies include 1) building a mouse model for Cystinosin7Δ to test if chemical chaperones can work in animal models; 2) working with chemists to optimize the current chemical chaperones; 3)identifying and studying other short-lived cystinosin mutants.
Lysosome biogenesis under the context of cancer development and progression
Most cancer cells are rapidly growing and dividing populations that require a lot of nutrients. Many of them can dramatically upregulate their autophagy and lysosomes to meet the high nutrient demands. For example, melanoma and pancreatic cancer cells can increase their lysosome numbers by up to 10-fold. How do cancer cells achieve such a remarkable upregulation? Is it through transcriptional stimulation? We are using a combination of genome-wide CRISPR knockout screening, cutting-edge live-cell imaging, and biochemical reconstitution to study this question. Inhibiting lysosome biogenesis in these cancer cells might shed light on developing new therapeutic strategies.