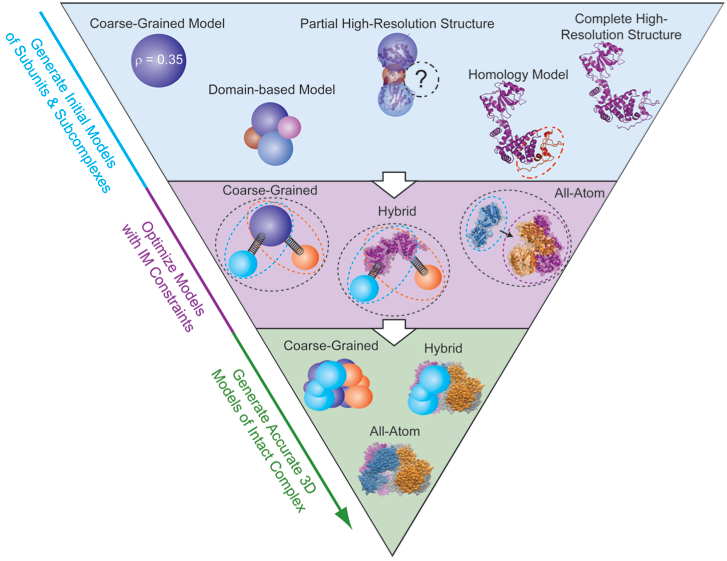
By combining the data generated by IM-MS with bioinformatics and molecular dynamics we are positioned to generate a full, low-resolution 3D architecture for protein complexes. First, each protein in the complex must have a representation in the model. When a subunit structure is unknown, it can be represented as a sphere with a radius corresponding to its IM derived size value. At the other end of the spectrum, if an atomic resolution model of the protein exists it can be integrated directly into the modeling algorithm or be added to the final model later to reduce computational costs. IM-MS restraints can be used to restrain distances, angles, and ultimately the shapes of intact complexes. We have developed a unique computational platform for generating and scoring models based on IM-MS inputs, and are working toward a new framework for benchmarking models against known structures and characterizing their resolution.
- Welcome Addison, Hanyu, and Ryan!
- Congratulations to Iliana!
- Congratulations to Dr. Rojas Ramirez and Dr. Parson!
- Congratulations to Dr. Han!
- New Publication from the Ruotolo Lab Featured in UM News!
Contact
Ruotolo Group
University of Michigan
Department of Chemistry
Room 4550
930 N. University Ave, Ann Arbor, MI 48109-1055
Phone: (+1) 734-763-2443
Fax: (1+) 734-615-3718